> Home > オートファジーの現状

オートファジー研究の現状と今後の展開
(MBL オートファジー総合カタログから転載(一部改変)
1.オートファジーとは
リソソームは分解を主たる役割とする細胞小器官である。エンドサイトーシス経路によって運ばれてくる細胞外成分や細胞膜タンパク質を分解する場所としてよく知られているが、細胞内成分を分解することももちろん可能である(図1)。オートファジーとは「細胞内の自己成分をリソソームで分解する細胞の機能」である。しばしば細胞死の一種と間違われることがあるが、あくまでも細胞内の分解機構の名前であり、多くの場合は細胞にとって保護的な作用をもつ。
オートファジーは「マクロオートファジー」、「ミクロオートファジー」、「シャペロン介在性オートファジー」に大別されている(図1)。このなかではマクロオートファジーがもっとも分解活性が高いと考えられており、酵母から動植物にいたる多くの生物で研究が進んでいる。一方、ミクロオートファジーは主に酵母で、シャペロン介在性オートファジーは主に哺乳動物で研究が進められているが、その分子実体や普遍性は解明途中にある(哺乳類ではミクロオートファジーと後期エンドソーム形成(多胞体)が同一の現象であるとする見方もある)。そのため、単にオートファジーと言う場合はマクロオートファジーを指すことがほとんどであり、本稿でもこれ以降そのように呼ぶことにする。
オートファジーに関わる分子群の多くは1990年代初頭の大隅良典博士(現・東京工業大学)らによる出芽酵母を用いた遺伝学的研究によって明らかにされた[1]。2016年8月現在、ATG1からATG41まで報告されている。これらの大半は限られた分解基質を対象とする選択的オートファジーに必要なものである。例えば、ATG30はペルオキシソームのオートファジー(ペキソファジー)、ATG32はミトコンドリアのオートファジー(マイトファジー)にのみ必要である。一方、飢餓で誘導される非選択的な「ふつうのオートファジー」などを含めたすべてのタイプのオートファジーに必要な「コアATG遺伝子」と呼ばれるものはATG1〜10、12〜14、16、18の15個である。これらは哺乳類を含めた他の生物でも高度に保存されている。その詳しい機能については他の総説を参照いただきたい[2, 3]。
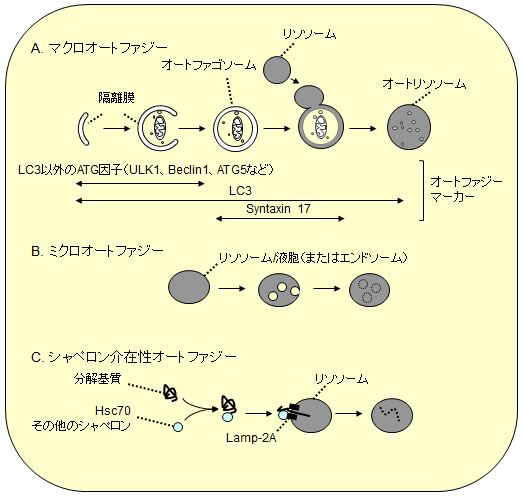 |
| 図1 3つのタイプのオートファジー |
| A.マクロオートファジー:直径約1 μmのオートファゴソームが形成されながら細胞質の一部を取り囲み、それがリソソームと融合するとオートファゴソームの内膜とともに内容物が分解される。LC3ファミリータンパク質はこの間のほぼすべての過程の膜に局在するため、オートファジー関連膜の全般的マーカーとして利用できる(オートリソソームになると内膜のLC3は分解され、外膜のLC3は膜からは徐々に外れる)。一方、LC3以外のほとんどのATG因子は閉鎖前の隔離膜にのみ局在し、リソソームとの融合に必要なシンタキシン17は閉鎖後のオートファゴソームに局在するので、それぞれの特異的マーカーとして利用できる。
|
2.オートファジー研究の現状
酵母のATG遺伝子の発見をきっかけに、オートファジーの研究は全く新しい展開を迎えることになった。それらには主に3つ流れがあると思われる。
第1は、酵母の優れた遺伝学を中心にした酵母Atg分子群の機能解析である。本家の大隅グループを世界の中心として、遺伝学、生化学、形態学、構造生物学などを駆使して、個々のAtgタンパク質や複合体の機能、それらの遺伝学的階層性、活性制御機構、基質認識機構などの幅広い範囲で理解が進んだ。酵母では、液胞近傍に原則としてひとつだけ存在する「前オートファゴソーム構造体(PAS)」からオートファゴソームが作られるが、その詳細がわかりつつある。しかし、オートファゴソーム膜の由来、膜のタンパク質や脂質組成、オートファゴソームの膜閉鎖機構、液胞との融合機構など、わからないこともまだ多くある。
第2は、酵母以外の生物でのATG相同分子の解析である。全ゲノムが解読される以前から、哺乳類や植物を含む多くの生物にATG相同分子が存在することは明らかであった。例外的に紅藻類や一部の原虫ですべてまたは一部が保存されていないことがわかっているが[4]、それらを除けばすべての真核生物でおおむね保存されていると言って良い。さらに高等動物では酵母には存在しないオートファジー必須因子(VMP1、EPG5、Ei24など)も発見されている。これらの発見は、オートファジー分子機構の解明に酵母と相補的に寄与してきたとともに、オートファジーの研究に欠かせないマーカーを提供し得た点においても重要である。吉森保博士(現・大阪大学)によって発見され、現在もっとも汎用されているオートファゴソームマーカーLC3Bなどがその良い例である(図1)[5, 6]。出芽酵母以外の多くの生物では、オートファゴソームは細胞内の複数の場所で同時多発的に作られる。これらの場所は、少なくても小胞体と強い関係にあることがわかっている。
第3は、逆遺伝学的手法を用いることによって、さまざまな生物でのatg欠損変異体の表現型解析がなされたことである。マウスでは単純ノックアウトマウスに加えて、多くのATG遺伝子の組織特異的ノックアウトマウスが作製された。筆者らの作製したATG5floxマウスだけでもすでに400以上の研究室に分与したので、おそらく一般に入手可能なすべてのCre発現マウスとは交配されたと思われる。その結果、オートファジーによる分解には基本的には2つの主要な役割があることがわかった[7]。一つは分解産物の供給であり、この機能や飢餓時や初期胚発生時の細胞内アミノ酸プールの維持や内因性抗原提示などに重要である。オートファジー不能酵母細胞が飢餓耐性をもたないということが最初の発見である[1]。この役割はすべての生物に共通するものであると考えられる。もうひとつの役割は分解による細胞内品質管理である。この目的には非選択的なバルクオートファジーとともに、基質選択的オートファジーが重要な働きをする。選択的基質としては、p62/SQSTM1などのLC3結合タンパク質、脱分極したミトコンドリアなどの損傷オルガネラ、細胞内侵入病原体(一部は細菌を取り囲む損傷エンドソームとして理解されている)などがあげられる。特に小松雅明博士(現・新潟大学)らによって明らかにされたように、p62/SQSTM1を低レベルに保つことは、凝集体の形成や、酸化ストレス応答性Nrf2経路の暴走などを抑制する上で重要である。総じて、これらの品質管理が不全となると、神経や肝をはじめとする細胞変性や加齢性変化の促進、腫瘍形成、感染の重症化がおこることがマウスで観察されている。これらの品質管理機能はタンパク質の平均半減期が細胞の倍加時間を大きく上回る場合(出芽酵母など)はほとんど問題にならず、寿命の長い細胞でより重要となる。マウス以外の多くの生物でも同様の生理学的研究がなされているが、紙面の都合上ここでは省略させていただく。
3.ヒト疾患とオートファジーについて
日本を中心としてオートファジーの基礎研究はこの10年あまりで劇的に進展したが、医学との関係の理解はまだ遙かに遅れていると言わざるを得ない。しかしながら、最近になってヒト遺伝学的研究からオートファジーとヒト疾患との関係が見えてきている。
まず2007年に炎症性腸疾患クローン病のリスクアレルとしてATG16L1が同定された[8, 9]。これは欠損変異ではなく、ATG16L1のほぼ中央に位置する300番目のトレオニンがアラニンに置換される変異(T300A)であり、健常人にも頻繁にみられる。ホモ変異でもクローン病発症のオッズ比は2倍程度である。また、この変異領域を含むC末端領域は酵母Atg16には存在せず、哺乳類においてもこの領域の機能はほとんどわかっていない。T300A変異に関してもオートファジー活性に影響を与えるとする報告と、与えないとする報告の両者がある。いずれにしても、多集団を対象とした統計学的解析ではじめて明らかにされた高頻度変異であるため、その意義を培養細胞や小数のマウスの実験で明らかにするのは容易ではないことが予想される。
次に2008年に米国のRichard Youleらによって、膜電位が低下したミトコンドリアのオートファジーによる分解(マイトファジーと呼ばれる)に、家族性パーキンソン病の原因遺伝子Parkin/PARK2が関与することが報告された[10]。Parkinはユビキチンリガーゼであり、脱分極した不良ミトコンドリアに局在する。この過程には家族性パーキンソン病の別の関連因子であるPINK1/PARK6とリン酸化されたユビキチンが必要であることがわかっている[11]。従って、ParkinやPINK1の変異を原因とするパーキンソン病はマイトファジーの不全によって本来分解されるべき不良ミトコンドリアが蓄積したために生じるものと考えられるようになった。しかし、Parkinにはミトコンドリアを温存したまま外膜タンパク質のみの分解を誘導するなど、マイトファジー以外の機能も有しているため[12]、マイトファジーの異常そのものが本当にパーキンソン病の原因であることを示すには生体内でのさらなる実験が必要であると考えられる。
さらに2012年以降、患者家系のエクソーム解析によって次々とオートファジー関連遺伝子の変異が見つかってきている。米国のHayflick博士らのグループと、松本直通博士・才津浩智博士(横浜市大)らのグループは独立してSENDA(static encephalopathy of childhood with neurodegeneration in adulthood)の原因として、WDR45/WIPI4遺伝子の変異を発見した[13, 14]。WIPI4は酵母ATG18またはATG21のヒトホモログ(WIPI1〜4が存在)の一つであり、コアATG遺伝子の欠損変異をもつヒト疾患の同定としては初めてとなった。WDR45はX染色体にコードされる遺伝子であり、患者のほとんどは女性のモザイクである。SENDA(現在はBPANとも呼ばれている)は大脳基底核などの脳内の鉄沈着を特徴とする神経変性疾患であり、小児期は非進行性の知的運動障害を呈し、20-30歳を過ぎることからパーキンソン病様症状が一気に進行する。患者リンパ芽球でオートファジー活性が低下することを筆者らは確認している[14]。しかし、WIPI4の正確な機能はまだ明確ではない。HeLa細胞なとの一般培養細胞ではWIPIファミリータンパク質のなかではWIPI2が圧倒的に重要である。それでも、最近作製されたWIPI4ノックアウトマウスの神経ではオートファジーの選択的基質であるp62の蓄積が観察されるので、神経系ではWIPI4がより重要な働きをしている可能性がある[15]。他のコアATG遺伝子変異としては、最近知的障害を伴う先天性失調症でATG5の変異が報告された[16]。これは、ATG12との結合不全による部分的なオートファジー活性の低下をもたらす。このほか、Vici症候群(脳梁無発育、白内障、心筋症、免疫不全、低色素症などを伴う)ではオートリソソーム分解に関わるEPG5遺伝子[17]、遺伝性痙性対麻痺でLC3結合タンパク質TECPR2の遺伝子[18]、小脳萎縮症でリソソームのPI(3,5)P2結合タンパク質SNX14の遺伝子[19]のそれぞれの変異がエクソーム解析によって明らかにされ、オートファジー活性が低下することも示されている。しかし、これらはオートファジー経路そのものというよりもリソソームの異常が主体であると考えられる。詳しくは他の総説を参照されたい[20]。
4.オートファジーを標的とした治療
オートファジーがヒト疾患の直接の原因になっているケースはまだ少ないと考えられるが、一方でオートファジーを治療標的にする試みはすでに始まっている。実際に臨床治験が行われている薬剤は、悪性腫瘍に対するクロロキン(またはハイドロキシクロロキン)である[20]。クロロキンはリソソーム機能を抑制する薬剤であり、必ずしもオートファジーを特異的ではないが、オートファジーも阻害するということで使用されている。米国ペンシルベニア大学を中心に多施設で臨床治験が進行中で、2016年8月現在、NIHのClinicalTrials.gov(https://clinicaltrials.gov/ct2/home)にはハイドロキシクロロキンは27件、クロロキンは8件がフェーズ1〜2として登録されている。多くは他剤との併用である。それらの結果の一部については、2014年8月にAutophagy誌に6編の詳細な論文として報告されており、すでに一部の患者で効果があるとのことである。なぜがんの治療にオートファジー阻害が有効であるのか諸説あるが、単なるアミノ酸産生としての機能だけではなく、より広範な細胞リモデリングや品質管理機構としてのオートファジー機能の阻害に起因する可能性もある[22, 23]。オートファジーに特に依存したがんが特定できれば、より効果的であると思われる。しかし、一方でクロロキンのオートファジー阻害以外の効果が抗がん作用を有しているという説も有り[24]、今後より特異性の高いオートファジー抑制剤の効果を検討する必要があろう。
一方で、まだ大規模な臨床治験には至っていないものの、神経変性疾患も有効な標的になりうると考えられている。神経変性疾患の多くは細胞内に異常なタンパク質が蓄積することが一因であると考えられている。そこで、オートファジーがもつ細胞内浄化作用を活性化することで、細胞にとって毒性をもつ異常タンパク質や変性タンパク質を除去することが試みられている。マウスやショウジョウバエなどの変性疾患モデル(ポリグルタミン病など)を用いた実験では、mTORC1阻害剤であるラパマイシンやその誘導体が症状の軽減に効果的であることが観察されている。しかし、mTORC1阻害剤は副作用が強いためにヒト疾患の治療には適していない。そこでmTORC1阻害を介さないオートファジー活性化剤がこれまでに探索されてきた。そのひとつは抗てんかん薬のカルバマゼピンである。実際に、肝細胞の小胞体内に変異タンパク質が蓄積することで肝障害を引き起こすα1アンチトリプシン欠損症のモデル動物に対してカルバマゼピンが有効であったとする報告がある[25]。この治療効果が真にオートファジー活性化を介したものであるのかどうかは不明であるが、今後期待されるアプローチである。
5.今後の課題と展開
オートファジーのメカニズム、生理的意義についてはこれまでかなりのことがわかってきており、まだ残されている重要課題が多くあるものの、今後着実に解明に向かって行くと予想される。一方、オートファジー関連因子がオートファジー以外の経路でも使用されていることがわかってきたことは新しい展開であろう。これには、オートファジー因子(ULK複合体を除く)がファゴソームの成熟を促進するとされる「LAP(LC3-associated phagocytosis)」や、オートファゴソームまたはその類似構造体が非典型的分泌に使われる経路などがあげられる[26, 27]。これらの生理的意義はまだ明らかにされつつある段階であるが、死細胞の貪食処理、自己免疫疾患、サイトカイン分泌などの免疫系との関連も深いと考えられている。
一方で、オートファジーの生体内でのモニター方法にはまだ課題が多い。培養細胞の基礎実験レベルであってもオートファジー活性測定法は未だに複雑で有り、十分に満足に行くものではない。例えば、オートファゴソーム数やLC3-IIフォーム(LC3の膜結合型)の増加は無条件にオートファジーの活性化を示すわけではなく、リソソームの阻害などの後半ステップのブロックを示している可能性もある[6]。実際、化合物スクリーニングなどでLC3の輝点やLC3-IIが増加する場合の多くは、オートファジー誘導では無く後半のブロックである。クロロキンに代表される弱塩基化合物の多くはこのような性質を持つ。最近ではこの注意点はかなり浸透し、より適切な方法である「フラックスアッセイ」が併用されるようになってきた。しかし、固定されたサンプルでフラックスアッセイを行うことは困難または不可能であり、病理組織学的解析における大きな障壁となっている。私たちは最近、培養細胞でも動物個体でも使用可能なオートファジーフラックスプローブを開発したが[28]、それでも遺伝子導入や臓器サンプル採取を必要とするので、生きている人間でオートファジー活性を測定するのは現時点ではほぼ絶望的であるといわざるを得ない。完璧とはいかなくても、ある程度オートファジー活性を推定できる間接的な方法を開発する必要があろう。それによってオートファジー活性が部分的に変化したヒト疾患なども明らかにされてくる可能性がある。現在のところ、オートファジー関連遺伝子の変異を見つける以外の方法でオートファジーの関与を知る機会は非常に限られている。
最後にオートファジー創薬であるが、こちらは十分に可能性があると予想している。よく、オートファジーを活性化することによる副作用がないかという懸念を聞くが、筆者はこの点は楽観的に考えている。まず、もともと低い定常レベルのオートファジーをノックアウトすることで異常タンパク質が顕著に増加することは、そのわずかなオートファジーが十分な細胞内浄化作用を持っていることを意味する。そうであれば、わずかな活性化でもある程度の効果が期待できると予想してもよいはずである。また、細胞内での分解と合成との間には精緻なフィードバックがあるはずで、分解の活性化は合成の活性化によって相補されることが予想される。すなわち、分解のみが活性化するのではなく、ターンオーバーが活性化すると考えれば、毒性はそれほどないであろう。むしろオートファジー経路だけをより特異的に活性化する薬剤を開発することが重要であると考えられる。
文献
1. Tsukada, M. and Y. Ohsumi, FEBS Lett., 1993. 333:169-174.
2. Nakatogawa, H., et al., Nat. Rev. Mol. Cell Biol., 2009. 10:458-67.
3. Mizushima, N., T. Yoshimori, and Y. Ohsumi, Annu. Rev. Cell Dev. Biol., 2011. 27:107-132.
4. Shemi, A., S. Ben-Dor, and A. Vardi, Autophagy, 2015. 11:701-15.
5. Kabeya, Y., et al., EMBO J., 2000. 19:5720-5728.
6. Mizushima, N., T. Yoshimori, and B. Levine, Cell, 2010. 140:313-26.
7. Mizushima, N. and M. Komatsu, Cell, 2011. 147:728-41.
8. Hampe, J., et al., Nat. Genet., 2007. 39:207-11.
9. Rioux, J.D., et al., Nat. Genet., 2007. 39:596-604.
10. Narendra, D., et al., J Cell Biol, 2008. 183:795-803.
11. Durcan, T.M. and E.A. Fon, Genes Dev., 2015. 29:989-999.
12. Scarffe, L.A., et al., Trends Neurosci., 2014. 37:315-24.
13. Haack, T.B., et al., Am. J. Hum. Genet., 2012. 91:1144-1149.
14. Saitsu, H., et al., Nat. Genet., 2013. 45:445-449.
15. Zhao, Y.G., et al., Autophagy, 2015. 11:881-90.
16. Kim, M., et al., Elife, 2016. 5: e12245.
17. Cullup, T., et al., Nat. Genet., 2013. 45:83-7.
18. Oz-Levi, D., et al., Am. J. Hum. Genet., 2012. 91:1065-1072.
19. Akizu, N., et al., Nat. Genet., 2015. 47:528-534.
20. Jiang, P. and N. Mizushima, Cell Res., 2014. 24:69-79.
21. Amaravadi, R.K., et al., Clin. Cancer Res., 2011. 17:654-66.
22. Cheong, H., et al., Nat. Biotechnol., 2012. 30:671-678.
23. White, E., Nat. Rev. Cancer, 2012. 12:401-410.
24. Maycotte, P., et al., Autophagy, 2012. 8:200-212.
25. Hidvegi, T., et al., Science, 2010. 329:229-32.
26. Bestebroer, J., et al., Traffic, 2013. 14:1029-41.
27. Ponpuak, M., et al., Curr. Opin. Cell Biol., 2015. 35:106-116.
28. Kaizuka, T., et al., Mol. Cell, in press
参考図書
水島昇、吉森保 編 「オートファジー: 生命をささえる細胞の自己分解システム (DOJIN BIOSCIENCE SERIES)」 化学同人 (2013)
水島昇 「細胞が自分を食べる オートファジーの謎」 PHPサイエンス・ワールド新書 2011年